

nejmoa2115869
Gilmar Reis, M.D., Ph.D., Eduardo A.S.M. Silva, M.D., Ph.D., Daniela C.M. Silva, M.D., Ph.D., Lehana Thabane, Ph.D., Aline C. Milagres, R.N., Thiago S. Ferreira, M.D., Castilho V.Q. dos Santos, Vitoria H.S. Campos, Ana M.R. Nogueira, M.D., Ana P.F.G. de Almeida, M.D., Eduardo D. Callegari, M.D., Adhemar D.F. Neto, M.D., Ph.D., et al., for the TOGETHER Investigators*
Abstract
BACKGROUND
The efficacy of ivermectin in preventing hospitalization or extended observation in an emergency setting among outpatients with acutely symptomatic coronavirus disease 2019 (Covid-19), the disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is unclear.
METHODS
We conducted a double-blind, randomized, placebo-controlled, adaptive platform trial involving symptomatic SARS-CoV-2–positive adults recruited from 12 public health clinics in Brazil. Patients who had had symptoms of Covid-19 for up to 7 days and had at least one risk factor for disease progression were randomly assigned to receive ivermectin (400 μg per kilogram of body weight) once daily for 3 days or placebo. (The trial also involved other interventions that are not reported here.) The primary composite outcome was hospitalization due to Covid-19 within 28 days after randomization or an emergency department visit due to clinical worsening of Covid-19 (defined as the participant remaining under observation for >6 hours) within 28 days after randomization.
RESULTS
A total of 3515 patients were randomly assigned to receive ivermectin (679 patients), placebo (679), or another intervention (2157). Overall, 100 patients (14.7%) in the ivermectin group had a primary-outcome event, as compared with 111 (16.3%) in the placebo group (relative risk, 0.90; 95% Bayesian credible interval, 0.70 to 1.16). Of the 211 primary-outcome events, 171 (81.0%) were hospital admissions. Findings were similar to the primary analysis in a modified intention-to-treat analysis that included only patients who received at least one dose of ivermectin or placebo (relative risk, 0.89; 95% Bayesian credible interval, 0.69 to 1.15) and in a per-protocol analysis that included only patients who reported 100% adherence to the assigned regimen (relative risk, 0.94; 95% Bayesian credible interval, 0.67 to 1.35). There were no significant effects of ivermectin use on secondary outcomes or adverse events.
CONCLUSIONS
Treatment with ivermectin did not result in a lower incidence of medical admission to a hospital due to progression of Covid-19 or of prolonged emergency department observation among outpatients with an early diagnosis of Covid-19. (Funded by FastGrants and the Rainwater Charitable Foundation; TOGETHER ClinicalTrials.gov number, NCT04727424. opens in new tab.)
Methods
TRIAL DESIGN AND OVERSIGHT
We conducted this randomized, adaptive platform trial for the investigation of the efficacy of repurposed treatments for Covid-19 among adult outpatients at high risk for hospitalization.10 The trial was designed and conducted in partnership with local public health authorities from 12 cities in Brazil in order to simultaneously test potential treatments for early Covid-19 with the use of a master protocol. A master protocol defines prospective decision criteria for discontinuing interventions for futility, stopping owing to superiority of an intervention over placebo, or adding new interventions. Interventions that have been evaluated in this trial thus far include hydroxychloroquine and lopinavir–ritonavir (both in protocol 1)11 and metformin, ivermectin administered for 1 day, ivermectin administered for 3 days, doxazosin, pegylated interferon lambda, and fluvoxamine (all in protocol 2), as compared with matching placebos. The full trial protocol with the statistical analysis plan has been published previously10 and is available with the full text of this article at NEJM.org.
The trial began recruitment for its first investigational groups on June 2, 2020. The evaluation that is reported here involved patients who had been randomly assigned to receive either ivermectin or placebo between March 23, 2021, and August 6, 2021. The initial trial protocol specified single-day administration of ivermectin, and we recruited 77 patients to this dose group. On the basis of feedback from advocacy groups, we modified the protocol to specify 3 days of administration of ivermectin. Here, we present data only on the patients who had been assigned to receive ivermectin for 3 days or placebo during the same time period. The full trial protocol was approved by local and national research ethics boards in Brazil and by the Hamilton Integrated Research Ethics Board in Canada. The CONSORT (Consolidated Standards of Reporting Trials) extension statement for adaptive design trials guided this trial report.12 All the patients provided written informed consent.
The trial was coordinated by Platform Life Sciences, and Cardresearch conducted the trial and collected the data. The first and last authors had full access to all the trial data and vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol. The funders had no role in the design and conduct of the trial; the collection, management, analysis, or interpretation of the data; the preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication. Ivermectin was purchased at full cost.
PATIENTS
On presentation to one of the trial outpatient care clinics, potential participants were screened to identify those meeting the eligibility criteria. Inclusion criteria were an age of 18 years or older; presentation to an outpatient care setting with an acute clinical condition consistent with Covid-19 within 7 days after symptom onset; and at least one high-risk criterion for progression of Covid-19, including an age of 50 years or older, diabetes mellitus, hypertension leading to the use of medication, cardiovascular disease, lung disease, smoking, obesity (defined as a body-mass index [the weight in kilograms divided by the square of the height in meters] of >30), organ transplantation, chronic kidney disease (stage IV) or receipt of dialysis, immunosuppressive therapy (receipt of ≥10 mg of prednisone or equivalent daily), a diagnosis of cancer within the previous 6 months, or receipt of chemotherapy for cancer. Patients who had been vaccinated against SARS-CoV-2 were eligible for participation in the trial. Further inclusion and exclusion criteria are listed in the trial protocol.10
If a patient met these eligibility criteria, trial personnel obtained written in-person informed consent and performed a rapid antigen test for SARS-CoV-2 (Panbio, Abbott Laboratories) to confirm eligibility for the trial. Before randomization, trial personnel obtained data on demographic characteristics, medical history, concomitant medications, coexisting conditions, and previous exposure to a person with Covid-19, as well as the score on the World Health Organization (WHO) clinical progression scale.13 Participants also completed the Patient-Reported Outcomes Measurement Information System (PROMIS) Global-10 health scale, which allows for the measurements of symptoms, functioning, and health-related quality of life (scores range from 5 to 20, with higher scores indicating better health-related quality of life). Normalized values are presented.
SETTING
The Supplementary Appendix, available at NEJM.org, lists the cities and investigators of the 12 participating clinical sites. Local investigators, in partnership with local public health authorities, recruited outpatients at community health facilities. Recruitment was supplemented by social media outreach.
RANDOMIZATION AND INTERVENTIONS
An independent pharmacist conducted the randomization at a central trial facility, from which the trial sites requested randomization by means of text message. Patients underwent randomization by means of a block randomization procedure for each participating site, with stratification according to age (<50 years or ≥50 years). The trial team, site staff, and patients were unaware of the randomized assignments. The active-drug and placebo pills were packaged in identically shaped bottles and labeled with alphabetic letters corresponding to ivermectin or placebo. Participants who were randomly assigned to receive placebo were assigned to a placebo regimen (ranging from 1 day to 14 days) that corresponded with that of a comparable active-treatment group in the trial. Only the pharmacist who was responsible for randomization was aware of which letter referred to which assignment.
All the patients received the usual standard care for Covid-19 provided by health care professionals in Brazil. Patients received either ivermectin at a dose of 400 μg per kilogram for 3 days or placebo beginning on the day of randomization, once per day. The placebos that were used in the trial involved regimens of 1, 3, 10, or 14 days in duration, according to the various comparator groups in the trial at the time of randomization. Patients were advised to take the pill on an empty stomach. Patients were shown a welcome video with information on the trial, ivermectin, adverse events, and follow-up procedures. Clinicians provided consultation on the management of symptoms and provided antipyretic agents; clinicians recommended antibiotic agents only if they suspected bacterial pneumonia.
OUTCOME MEASURES
The primary composite outcome was hospitalization due to Covid-19 within 28 days after randomization or an emergency department visit due to clinical worsening of Covid-19 (defined as the participant remaining under observation for >6 hours) within 28 days after randomization. Because many patients who would ordinarily have been hospitalized were prevented from admission because of limited hospital capacity during peak waves of the Covid-19 pandemic, the composite outcome was developed to measure both hospitalization and a proxy for hospitalization, observation in a Covid-19 emergency setting for more than 6 hours. This region of Brazil implemented mobile hospital-like services in the emergency settings (i.e., temporary field hospitals) with units of up to 80 beds; services included multiple-day stays, oxygenation, and mechanical ventilation. The 6-hour threshold referred only to periods of time that were recommended for observation by a clinician and was discounted for wait times. The event-adjudication committee, whose members were unaware of the randomized assignments, judged the reason for hospitalization or prolonged observation in the emergency department as being related or unrelated to the progression of Covid-19. Guidance for the validity of composite outcomes indicates that outcomes should have a similar level of patient importance.14
Secondary outcomes included SARS-CoV-2 viral clearance at day 3 and day 7, as assessed with the use of the quantitative reverse transcriptase–polymerase chain reaction laboratory test kit for SARS-CoV-2 from Applied Biosystems; hospitalization for any cause; the time to hospitalization; the duration of hospitalization; the time to an emergency visit lasting more than 6 hours; the time to clinical recovery, as assessed with the use of the WHO clinical progression scale13; death from any cause; the time to death; receipt of mechanical ventilation; the number of days with mechanical ventilation; health-related quality of life, as assessed with by the PROMIS Global-10 physical score and mental health score; the percentages of patients who adhered to the assigned regimen; and adverse reactions to ivermectin or placebo. We assessed all the secondary outcomes through 28 days after randomization.
TRIAL PROCEDURES
Trial personnel obtained outcome data by means of in-person, telephone, or WhatsApp (a smartphone app for video-teleconferencing) contact on days 1, 2, 3, 4, 5, 7, 10, 14, and 28. All the trial procedures are listed in the protocol. Adverse events were recorded at each participant contact date and were graded according to the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events.15 All serious and nonserious adverse events were reported to trial personnel according to local regulatory requirements. Reportable adverse events included serious adverse events, adverse events that resulted in the discontinuation of ivermectin or placebo, and adverse events that were assessed by the investigators as being possibly related to ivermectin or placebo.
DATA AND SAFETY MONITORING COMMITTEE OVERSIGHT
The data and safety monitoring committee met four times after the enrollment of the first patient to assess the probability of the superiority of ivermectin to placebo with regard to the primary outcome, on the basis of prespecified thresholds in the statistical analysis plan. On August 5, 2021, the data and safety monitoring committee recommended stopping the enrollment of patients into the ivermectin group because the planned sample size had been reached.
STATISTICAL ANALYSIS
The adaptive design trial protocol and the master statistical analysis plan (available with the protocol) provide details of the sample-size calculation and statistical analysis, including adapted approaches to sample-size reassessment.10 In planning for the trial, we assumed a minimum clinical utility of 37.5% of ivermectin (relative risk difference vs. placebo) in order for the trial to have 80% power, at a two-sided type I error of 0.05, for a pairwise comparison with placebo assuming that 15% of the patients in the placebo group would meet the primary outcome. This calculation resulted in a planned enrollment of 681 patients in each group.
Interim analyses were planned to occur after 25%, 50% and 75% of the maximum number of patient outcomes had been observed, as well as at the trial completion. The posterior efficacy threshold was set at 97.6% and the futility thresholds at 20%, 40% and 60%. If the intervention group showed a posterior probability of efficacy by crossing a boundary, it was to be stopped. These superiority and futility thresholds were determined on the basis of 200,000 simulation runs in which different values of the relative risk difference were considered (0, 20, and 37.5 percentage points).
The characteristics of the patients at baseline are reported as counts and percentages or, for continuous variables, as medians with interquartile ranges. We applied a Bayesian framework to assess the effect of ivermectin as compared with placebo on the primary outcome analysis and for the analyses of secondary outcomes. Posterior probability for the efficacy of ivermectin with regard to the primary outcome was calculated with the use of the beta-binomial model for the percentages of patients with an event, starting with uniform prior distributions for the percentages. Missingness in covariate data was handled with multiple imputation by chained equations.16
The intention-to-treat population included all the patients who had undergone randomization. The modified intention-to-treat population included all the patients who received ivermectin or placebo for at least 24 hours before a primary-outcome event (i.e., if an event occurred before 24 hours after randomization, the patient was not counted in this analysis). The per-protocol population included all the patients who reported 100% adherence to the assigned regimen. Although all the participants who had been assigned to the 3-day and 14-day placebo regimens were included in the intention-to-treat population, only those who had been assigned to the 3-day placebo regimen were included in the per-protocol population. The primary outcome was also assessed in subgroups defined according to participant age, body-mass index, status of having cardiovascular disease or lung disease, sex, smoking status, and time since symptom onset.
Secondary outcomes were assessed with the use of a Bayesian approach; given the Bayesian framework of our analysis, we did not test for multiplicity. We assessed time-to-event outcomes using Bayesian Cox proportional-hazards models, binary outcomes using Bayesian logistic regression, and continuous outcomes using Bayesian linear regression. Cause-specific Bayesian competing-risks survival analysis, with adjustment for death, was used for the time-to-recovery analysis. Per-protocol analyses were considered to be sensitivity analyses for the assessment of the robustness of the results. Personnel at Cytel performed all the analyses using R software, version 4.0.3. Further details are provided in the statistical analysis plan, which is available with the protocol.
Results
TRIAL POPULATION
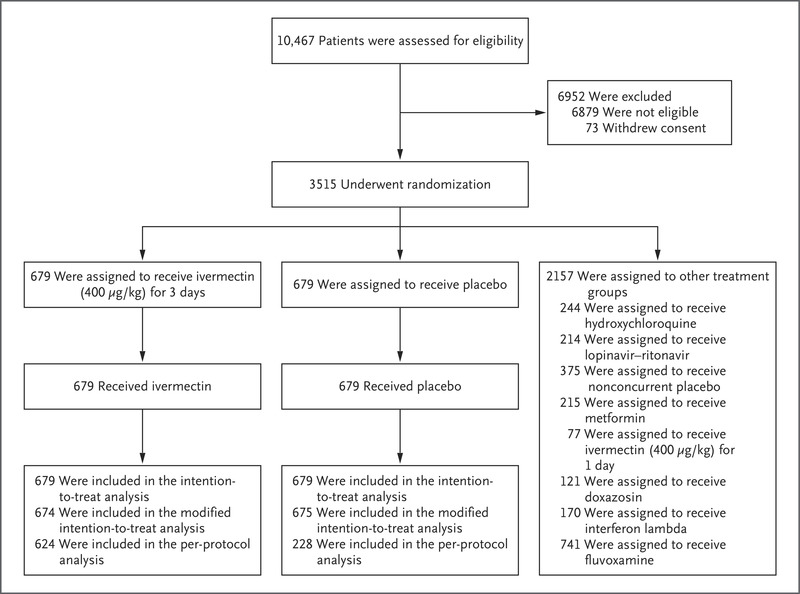 Randomization and Follow-up of the Patients.
Randomization and Follow-up of the Patients.
A total of 10,467 outpatients were screened for inclusion in the trial. Of these, 1358 patients were randomly assigned to receive either ivermectin (679 patients) or placebo (679 patients); 2157 patients were randomly assigned to other intervention groups (Figure 1). Here we report the findings for the evaluation of ivermectin as compared with placebo.
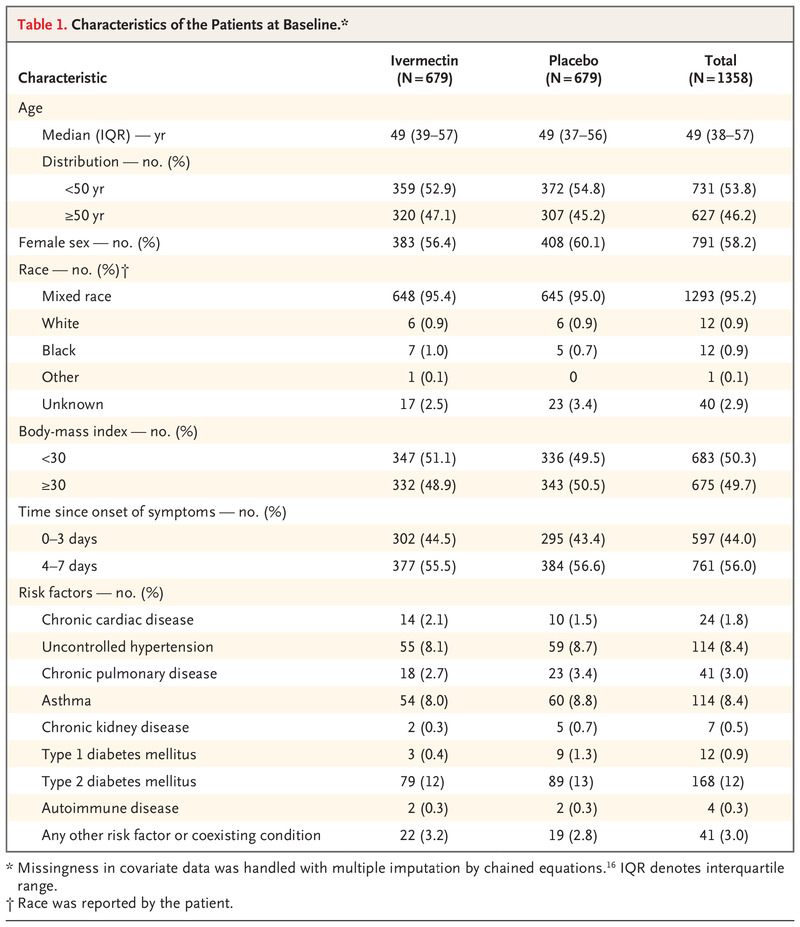 Characteristics of the Patients at Baseline.
Characteristics of the Patients at Baseline.
The median age of the patients was 49 years (interquartile range, 38 to 57), and 791 patients (58.2%) were women. Most of the patients identified as being of mixed race (1293 [95.2%]), with 12 patients (0.9%) identifying as White and 12 (0.9%) as Black. With respect to covariates, the data in Table 1 suggest that the groups were generally well balanced. The mean (±SD) number of days with Covid-19 symptoms before randomization was 3.8±1.9.
PRIMARY OUTCOME
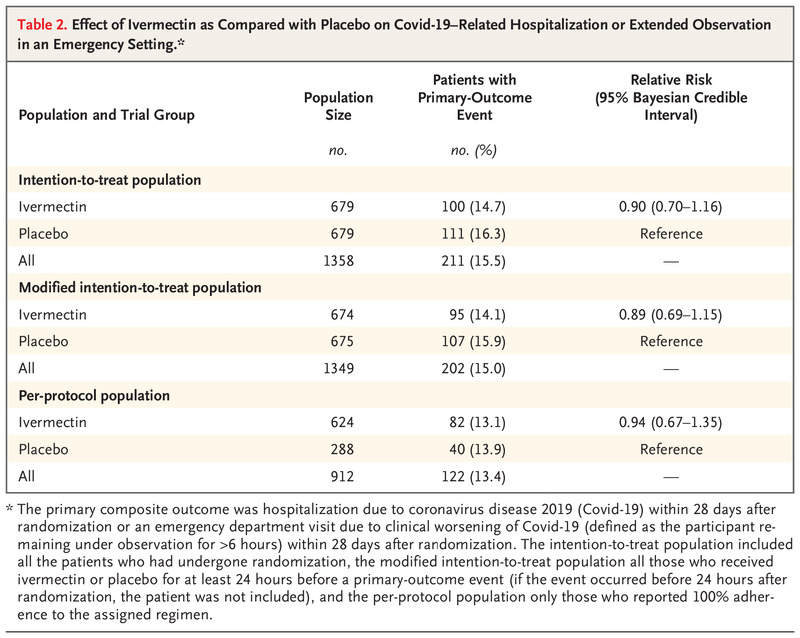 Effect of Ivermectin as Compared with Placebo on Covid-19–Related Hospitalization or Extended Observation in an Emergency Setting.
Effect of Ivermectin as Compared with Placebo on Covid-19–Related Hospitalization or Extended Observation in an Emergency Setting.
In the intention-to-treat population, 100 patients (14.7%) in the ivermectin group had a primary-outcome event (composite of hospitalization due to the progression of Covid-19 or an emergency department visit of >6 hours that was due to clinical worsening of Covid-19), as compared with 111 (16.3%) in the placebo group (relative risk, 0.90; 95% Bayesian credible interval, 0.70 to 1.16) (Table 2). Similar results were observed in the modified intention-to-treat population (relative risk, 0.89; 95% Bayesian credible interval, 0.69 to 1.15) and the per-protocol population (relative risk, 0.94; 95% Bayesian credible interval, 0.67 to 1.35). The probability that the percentage of patients with a primary-outcome event was lower in the ivermectin group than in the placebo group did not meet the prespecified threshold for superiority in any of these three trial populations. These posterior efficacy values were lower than the prespecified 97.6% threshold that had been set for the fourth and final interim analysis. Of all the primary-outcome events in the intention-to-treat population, most events were hospitalizations (171 of 211 events [81.0%]). Primary-outcome events in the trial happened a median of 5 days (interquartile range, 3 to 7) after randomization. Data for the components of the primary outcome are shown in Table S1 in the Supplementary Appendix.
SECONDARY OUTCOMES
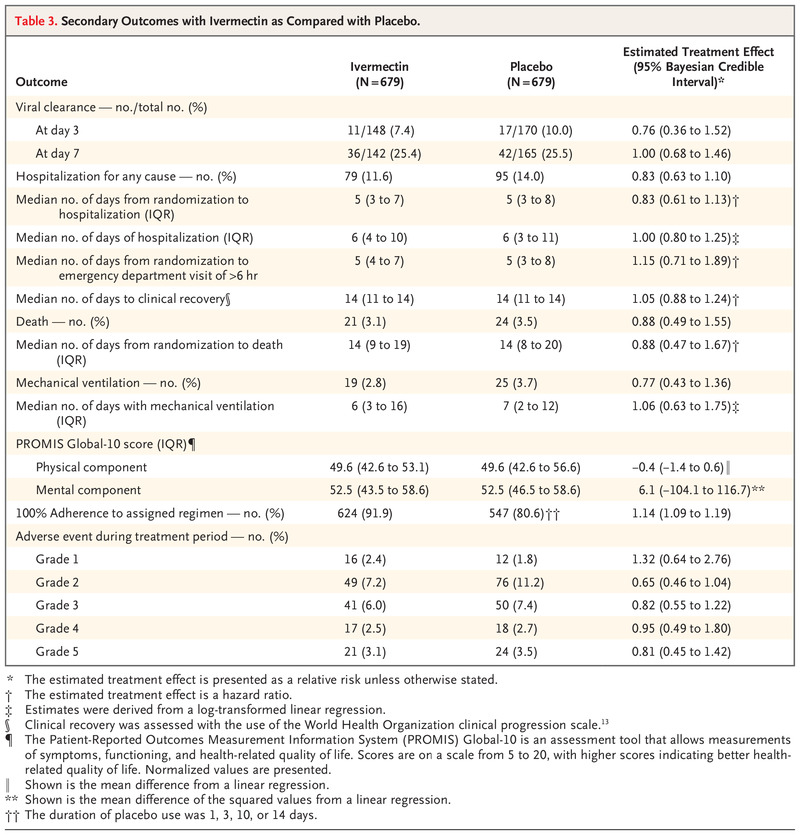 Secondary Outcomes with Ivermectin as Compared with Placebo.
Secondary Outcomes with Ivermectin as Compared with Placebo.
Table 3 presents findings of the secondary outcome analyses. There were no significant differences between the ivermectin group and the placebo group with regard to viral clearance at day 7 (relative risk, 1.00; 95% Bayesian credible interval, 0.68 to 1.46) (Fig. S3). The 14-day restricted mean survival time difference17 was 0.11 days (95% Bayesian credible interval, −0.23 to 0.48). There were no significant between-group differences with regard to the risk of hospitalization for any cause (relative risk, 0.83; 95% Bayesian credible interval, 0.63 to 1.10), the time to hospitalization (hazard ratio, 0.83; 95% Bayesian credible interval, 0.61 to 1.13) (Fig. S1), and the number of days in the hospital (mean difference of the log-transformed values, 1.00 days; 95% Bayesian credible interval, 0.80 to 1.25).
There were also no significant between-group differences in the time to clinical recovery (Fig. S2) (hazard ratio, 1.05; 95% Bayesian credible interval, 0.88 to 1.24), the risk of death (relative risk, 0.88; 95% Bayesian credible interval, 0.49 to 1.55), the time to death (hazard ratio, 0.88; 95% Bayesian credible interval, 0.47 to 1.67), or the number of days with mechanical ventilation (mean difference, 1.06 days; 95% Bayesian credible interval, 0.63 to 1.75). There was no evidence of between-group differences in the PROMIS Global-10 physical-component score as measured on day 28 (mean difference, −0.4 points; 95% Bayesian credible interval, −1.4 to 0.6) or mental-component score (mean difference of the squared values, 6.1 points; 95% Bayesian credible interval, −104.1 to 116.7). With regard to adverse events, there were no important between-group differences in the incidence of adverse events during the treatment period (Table S6).
SUBGROUP ANALYSES
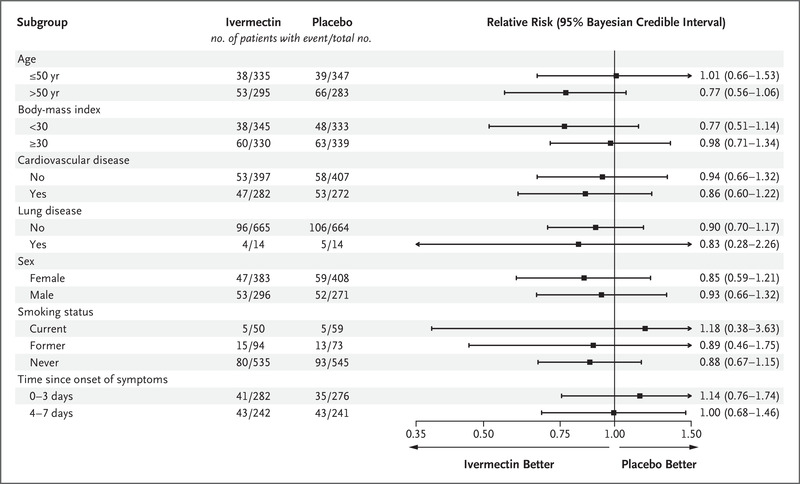 Subgroup Analyses of Ivermectin as Compared with Placebo for the Prevention of Covid-19–Related Hospitalization or Prolonged Observation in an Emergency Setting.
Subgroup Analyses of Ivermectin as Compared with Placebo for the Prevention of Covid-19–Related Hospitalization or Prolonged Observation in an Emergency Setting.
In prespecified subgroup analyses, there was no evidence of a treatment effect with ivermectin as compared with placebo in subgroups defined according to patient age, body-mass index, status of having cardiovascular disease or lung disease, sex, smoking status, or time since symptom onset (Figure 2 and Table S2). We observed no benefit with ivermectin as compared with placebo among patients who began the trial regimen within 3 days after symptom onset (relative risk, 1.14; 95% Bayesian credible interval, 0.76 to 1.74).
Discussion
We did not find a significantly or clinically meaningful lower risk of medical admission to a hospital or prolonged emergency department observation (primary composite outcome) with ivermectin administered for 3 days at a dose of 400 μg per kilogram per day than with placebo. We found no important effects of treatment with ivermectin on the secondary outcomes.
The evidence supporting the role of ivermectin in the treatment of Covid-19 is inconsistent. At least three meta-analyses of ivermectin trials have strongly indicated a treatment benefit, and others have concluded that there was no benefit.7,8,18-20 Although the number of included trials involving outpatients varies among the meta-analyses, the overall number of events that occurred in our trial is larger than the number of all the combined events in these meta-analyses. The results of this trial will, therefore, reduce the effect size of the meta-analyses that have indicated any benefits. In addition, a reported trial of ivermectin treatment for Covid-19 was suspected of malfeasance and was withdrawn from publication,9 and other trials have been weakened by concerns about quality.8 A large collaboration of clinical trialists working on ivermectin treatment for Covid-19 has conducted a meta-analysis of trials and has concluded that ivermectin did not offer a treatment benefit when trials that were considered to be of moderate or better quality were examined.6 The WHO has concluded, on the basis of results obtained before our trial, that there existed only very-low-certainty evidence regarding ivermectin and thus recommended against the use of ivermectin for the treatment of patients with Covid-19 outside the clinical trial setting.21 The findings in our trial are consistent with these conclusions.
Major strengths of our trial include the rapid recruitment and enrollment of high-risk patients. We enrolled only patients who had a confirmed diagnosis of Covid-19 and less than 7 days of symptoms, and we did not enroll asymptomatic SARS-CoV-2–positive persons. The primary outcome was a composite of hospitalization for adjudicated Covid-19 as well as retention in a Covid-19 emergency setting for physician observation for more than 6 hours. Patients in these settings would typically have been hospitalized but were prevented from doing so because of limited capacity in hospitals.
When we began this trial, we randomly assigned patients to receive a 1-day dose of ivermectin, as is most commonly used for the treatment of parasitic diseases. We responded to feedback from advocacy groups regarding this administration schedule and adapted the duration of ivermectin administration to 3 days at a relatively high dose as compared with most other trials of this drug. Ivermectin has been used off-label widely since the original in vitro study by Caly et al. describing ivermectin activity against SARS-CoV-2,22 and in Brazil, in particular, the use of ivermectin for the treatment of Covid-19 has been widely promoted. We ensured that trial participants did not have a history of ivermectin use for the treatment of Covid-19 by means of extensive screening of potential participants about this issue. Given the public interest in ivermectin and the support of its use by paramedical groups, we suspect that there will be additional criticism that our administration regimen was inadequate. Details of the pharmacologic rationale and mechanistic hypotheses for ivermectin use are provided in the Supplementary Appendix.
In this randomized trial, the administration of ivermectin did not result in a lower incidence of medical admission to a hospital or prolonged emergency department observation for Covid-19 among outpatients at high risk for serious illness.
Supplementary Material
| Protocol | 8478KB | |
| Supplementary Appendix | 682KB | |
| Disclosure Forms | 416KB | |
| Data Sharing Statement | 71KB |